
FDA und EU erweitern Abkommen über die gegenseitige Anerkennung erneut!

Am 31. Mai 2023 haben die U.S. Food and Drug Administration (FDA) und die Europäische Union (EU) die Erweiterung des Sektoralen Anhangs für die gute Herstellungspraxis (GMP) des US-EU-Abkommens über die gegenseitige Anerkennung (MRA) angekündigt. Diese Erweiterung umfasst nun auch die Inspektion von Tierarzneimitteln, die auch als "Tierarzneimittel" bezeichnet werden.

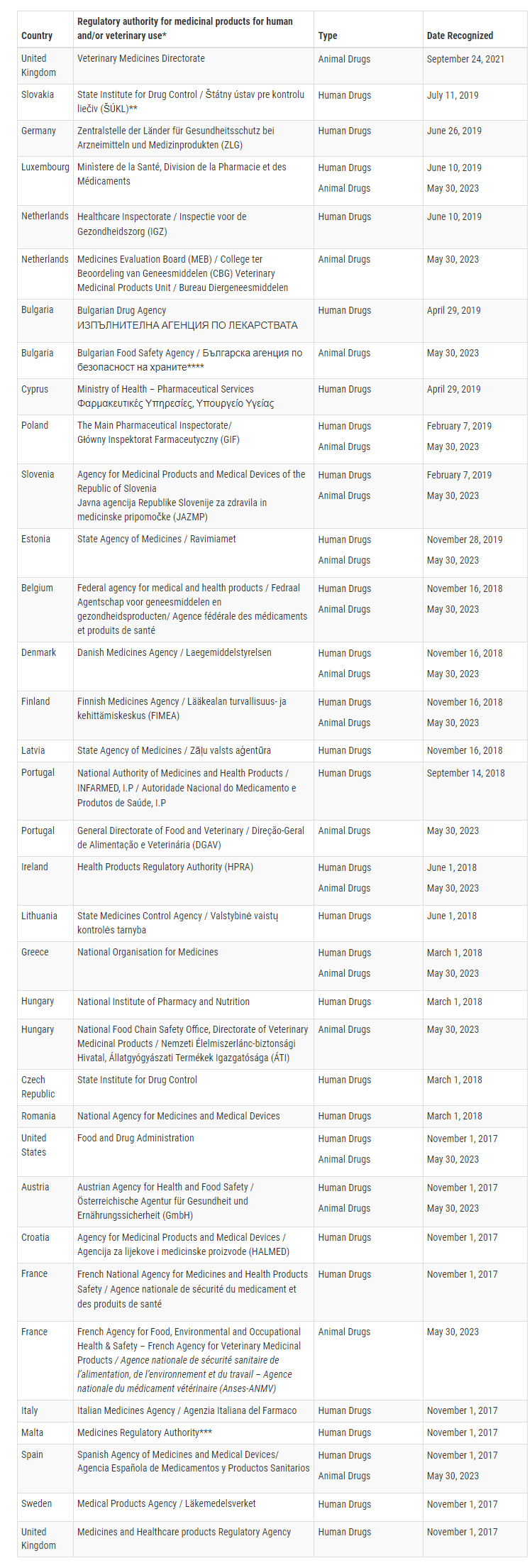
Derzeit hat die FDA bestätigt, dass 16 Mitgliedstaaten der Europäischen Union in der Lage sind, GMP-Inspektionen für bestimmte Tierarzneimittel auf einem Niveau durchzuführen, das dem der Vereinigten Staaten entspricht. Diese Mitgliedstaaten sind Belgien, Bulgarien, Dänemark, Estland, Finnland, Frankreich, Griechenland, Irland, Luxemburg, die Niederlande, Österreich, Polen, Portugal, Slowenien und Spanien.
Mit dieser erweiterten MRA kann sich die FDA auf GMP-Inspektionen verlassen, die von diesen 16 Mitgliedstaaten für Veterinärprodukte durchgeführt werden, während sich die Europäische Union auf Inspektionen durch die FDA verlassen wird. Diese gegenseitige Anerkennung verbessert die regulatorische Zusammenarbeit und strafft die Aufsicht über Tierarzneimittel zwischen der FDA und der EU.

Was ist MRA?
Vereinbarungen über die gegenseitige Anerkennung (Mutual Recognition Agreements, MRAs) erleichtern die Zusammenarbeit zwischen der FDA und ausländischen Aufsichtsbehörden und ermöglichen es Arzneimittelinspektoren, sich auf Inspektionsinformationen zu verlassen, die in den Gerichtsbarkeiten des jeweils anderen Landes durchgeführt werden. Der 2012 eingeführte Food and Drug Administration Safety and Innovation Act erteilt der FDA die Befugnis, Vereinbarungen zur Anerkennung von Arzneimittelinspektionen zu treffen, die von ausländischen Aufsichtsbehörden durchgeführt werden und die US-Anforderungen erfüllen.
MRAs dienen zwei Hauptzwecken:
1. Effizienzsteigerung: Durch die Vermeidung von Doppelinspektionen fördern MRAs optimierte Prozesse sowohl für US-amerikanische als auch für ausländische Regulierungssysteme.
2. Ressourcenzuweisung: MRAs ermöglichen die Umverteilung von Ressourcen, um sich auf die Inspektion von Arzneimittelherstellungsanlagen mit potenziell höheren Risiken für die öffentliche Gesundheit weltweit zu konzentrieren.

Wer hat MRAs mit der FDA unterzeichnet?
Derzeit verfügt die FDA über MRAs mit der Europäischen Union (EU) und dem Vereinigten Königreich (UK). Darüber hinaus unterzeichnete die FDA am 12. Januar 2023 ein MRA mit der Schweiz. Bevor diese MRA jedoch in Kraft tritt, muss die FDA die Inspektionskapazitäten der Schweiz bewerten, um sicherzustellen, dass sie den US-Anforderungen entsprechen. Ebenso muss das Schweizerische Heilmittelinstitut (Swissmedic) die Einhaltung der Schweizer Anforderungen durch die FDA beurteilen.

Einschränkungen
Die Fähigkeitsbeurteilungen beziehen sich derzeit auf routinemäßige Überwachungsinspektionen. Es besteht jedoch die Möglichkeit, dass die folgenden Produkt- und Inspektionstypen nach einer weiteren Bewertung in den Geltungsbereich der Vereinbarung aufgenommen werden:
1. Impfstoffe für den menschlichen Gebrauch
2. Aus Plasma gewonnene Pharmazeutika
3. Prüfpräparate (Material für klinische Prüfungen), die für jede Vereinbarung spezifisch sind
Die FDA und das Vereinigte Königreich haben die Möglichkeit erörtert, den Geltungsbereich der MRA auf Impfstoffe und aus Plasma gewonnene Arzneimittel für den menschlichen Gebrauch auszuweiten. Eine Entscheidung in dieser Angelegenheit wird im Juli 2025 nach Durchführung zusätzlicher Bewertungen erneut getroffen.
In ähnlicher Weise haben die FDA und die EU auch die Ausweitung des MRA auf Impfstoffe und aus Plasma gewonnene Arzneimittel für den menschlichen Gebrauch geprüft. Die FDA wird dieses Problem im Juli 2025 unter Berücksichtigung der Ergebnisse weiterer Bewertungen neu bewerten.
Ausgenommen vom MRA-Anwendungsbereich sind: Arzneimittel für neuartige Therapien (ATMPs), menschliches Blut, menschliches Plasma, menschliche Gewebe und Organe, Zellen (Swissmedic MRA) und veterinärmedizinische Immunologika.
PS: Wenn Sie möchten, dass unser Team ein neues Homecare-Produkt mit FDA, MDR, ISO, FSC und mehr anpasst, wenden Sie sich bitte an insights@medasiagroup.com
WIR EMPFEHLEN



Verwandte Beiträge




- MedInsights abonnieren
- MedInsights abonnieren
- MedInsights abonnieren
- MedInsights abonnieren
- MedInsights abonnieren